我们的服务
我们的服务 SERVICE
-
医疗器械注册
-
全球
-
西欧
-
北美
-
东亚
-
东盟
-
拉美
-
阿拉伯国家
-
南亚
-
澳洲
-
东欧
-
非洲
联系我们 LINK US
服务热线
400-888-1034
深圳亿联检测有限公司
0755-8936 6579
kefu@yi-link.cn
深圳市坪山区锦绣中路19号美讯科技园A1406

扫描关注亿联检测微信公众号
美国FDA认证介绍

美国联邦法典第21卷(即《联邦食品药品化妆品法案》),明确所有在美国市场销售的医疗器械必须向美国食品药品管理局(FDA)进行申报注册。业内把该过程通称为FDA认证。
FDA全称是Food and Drug Administration,是美国人类和健康服务部(Department of Health & Human Services,HHS)的下设机构之一,负责对药品、食品、化妆品、医疗器械、兽药等产品进行全面监督管理,其负责医疗器械的部门是CDRH(Center for Devices and Radiological Health)。
CDRH属下有7个办公室,其中器械评估办公室ODE(Office of Device Evaluation)有6个部门,这6个部门负责对所有医疗器械进行上市审批工作。
6个部门具体如下:
1.临床试验器械部;
2.常规、康复和神经科用器械部;
3.生殖、腹部和放射学用器械部;
4.心血管和呼吸用器械部;
5.牙科、传染病控制和普通医院器械部;
6.眼科和耳鼻喉科用器械部。
《联邦食品药品化妆品法案》把医疗器械分为三类,分别为:
Ⅰ类(一类产品):“普通控制(General Controls)”产品。是指危险性小或基本无危险性的产品,它的设计通常比Ⅱ类产品、Ⅲ类产品简单。例如医用手套、压舌扳、手动手术器械、温度计等,这类产品约占全部医疗器械品种的25%。
“普通控制”要求的具体规定是:
1.登记每一处生产场地;
2.列出已经进入市场的器械品种;
3.在销售新的器械或经过重要改造的器械之前提交“上市前通知”;
4.生产过程应符合GMP法规。
Ⅱ类(二类产品):”普通+特殊控制(General & Special Controls)”产品。Ⅱ类是指那些用普通控制不足以控制其安全性和有效性,必须通过现有的其他方式,即特殊控制,来保证其安全性和有效性的产品。
“特殊控制”规定是指除具备“普通控制”的要求外,申报单位还应提供正式颁布的标准、上市后监控的文件、疗效反馈登记、上市前的(临床试验)临床研究报告(包括临床和非临床的研究)等。特殊标签要求、强制性性能指标、售后监控都属于特殊控制方式。
常见的II类产品有心电图仪、超声诊断仪、输血输液器具、呼吸器等,II类产品约占全部医疗器械品种的55%。
Ⅲ类(三类产品)是指那些仅用普通控制和特殊控制还不足以确保其安全性和有效性的产品。因此,Ⅲ类除应符合“普通控制”和“特殊控制”的要求以外,还要提交针对预期医疗作用效果的证明文件,以及微生物、毒性、免疫、生物相容性、储存期限等的动物实验、临床研究报告。
Ⅲ类产品具有较大危险性或危害性,它通常用来支持人体生命,防止人体健康受损,具有致病、致残的潜在的、不合理的风险。例如人工心脏瓣膜、心脏起搏器、人工晶体、人工血管等,这类产品约占20%。
除非法规豁免,Ⅰ类、Ⅱ类器械及少量Ⅲ类器械通常要求上市前通知(Pre-market Notification);而Ⅲ类器械则大多数要求获得上市前批准(PMA)。
上市前通知业内通称为510(k),意在证明该产品与已经合法上市的产品实质性等同(Substantially Equivalent)。实质性等同是指:与已上市的产品预期用途相同;产品的新特性不会对安全性或有效性产生影响,或者对安全有效性产生影响的新特性有可接受的科学方法用于评估新技术的影响以及有证据证明这些新技术不会降低安全性或有效性。
品牌商必须在产品上市前90天向FDA提交申请,FDA审查该产品是否与已上市产品实质性等同。通过510(k)审查后,产品才可以在市场上销售。
上市前批准(PMA),意在提供足够、有效的证据证明医疗器械按照设计和生产的预期用途,能够确保产品的安全有效。
品牌商必须在产品上市前向FDA提交PMA申请书及相关资料,证明产品质量符合要求,临床使用安全、有效。FDA在收到PMA申请后45天内通知厂家是否立案审查,并在180天内对其做出是否批准的决定。
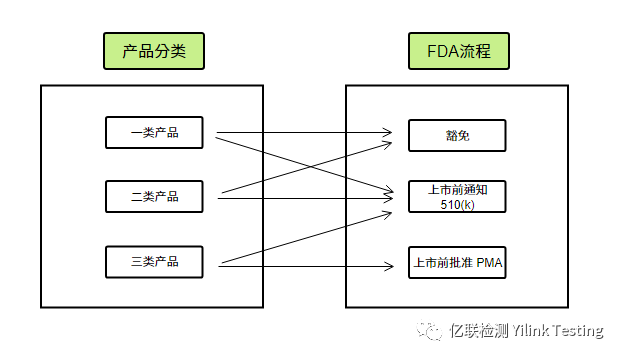
FDA产品分类和申请流程对照
需要强调的是,医疗器械的上市前审批由FDA进行统一管理,虽然有一部分产品可由第三方机构进行审评,但最终的批准权都还是在FDA。
在医疗器械完成豁免程序、510(k)申请或者PMA批准之后,即可以上市销售。上市销售的产品,依然受到FDA的强制性监管。
FDA通过对企业进行质量体系检查来进行上市后监督。对Ⅱ、Ⅲ类产品每两年检查一次质量体系,I类产品每四年检查一次质量体系。若存在隐患或发现问题,FDA随时可对企业进行检查。
FDA也全面负责医疗器械不良事件的监测和再评价工作。根据FDA规定,对于由医疗器械引起、可能引起或促使的死亡、严重伤害事件,不论医疗器械用户、经销商、制造商,都必须尽快报告。
FDA对发现的违规行为,会实施相应的行政处罚,其手段包括:发警告信、对伪劣或假冒产品行政扣押、对违法公司提起诉讼、召回产品等。召回产品可由FDA律师向法院申请强制执行。
亿联检测拥有丰富的医疗器械FDA合规经验,在I类产品、II类产品的510(k)申请上拥有众多成功案例。我们的医疗器械注册专家能够提供专业周到的服务,帮助中国品牌商和生产厂顺利进入美国市场。
注意事项
1.FDA 注册和上市外部链接免责声明并不表示公司或其设备的批准、许可或授权。 2.FDA 不会向医疗器械机构颁发注册证书,也不会对已注册其机构并列出其医疗产品的机构的信息进行认证。 3.FDA 标志仅供 FDA 官方使用,不适用于私营部门的材料。在私营部门材料上使用 FDA 标志可能会传达 FDA 支持或认可私营部门组织或该组织的活动、产品、服务和/或人员(公开或默认)的信息,而 FDA 没有也不能这样做。 4.需要注意的是,医疗器械产品的管理类别并不是一成不变的,随着与医疗器械有关的知识和经验的增长,产品的管理类别可以通过重新分类(reclassification)程序进行调整。管理类别的改变以FDA掌握的最新医疗器械信息为基础,FDA可以自发地或根据外界请求按照有关法律法规对医疗器械重新分类。 如果企业要求将自己生产的产品重新分类到较低的管理类别,就必须向FDA提供强有力的证明材料,证明该产品划分到较低的管理类别足以保证该产品的安全性和有效性。在对该产品管理类别的重新分类作出最终决定之前,FDA会在联邦登记上发布该产品重新分类的推荐性的规则,包括重新分类的科学判断,并请求公众参与评论。接着,才在联邦登记上公布该产品重新分类的最终决定。 申请流程 申请认证请联系亿联检测业务人员。
上一个: 北美安规认证标识
下一个:
联系我们
CONTACT US
您好,如有任何问题请联系我们,欢迎提交任何关于我们的问题和建议,我们将尽快回复您。 感谢您对我们的帮助。我们7*24小时竭诚为您服务!
-

kefu@yi-link.cn
-

深圳市坪山区锦绣中路19号美讯科技园A1406
-

0755-8936 6579
扫描关注亿联检测微信公众号